.png)

Autor: Dinler Amaral Antunes é professor assistente de Biologia Computacional no Departamento de Biologia e Bioquímica da Universidade de Houston, membro do Centro para Receptores Nucleares e Sinalização Celular (CNRCS), e da Sociedade Internacional de Biologia Computacional (ISCB).
O ano de 2020 vai ficar marcado pelos impactos da pandemia de COVID-19, mas também pela velocidade e dimensão da resposta gerada pela comunidade científica. O primeiro surto de disseminação do novo coronavirus (SARS-CoV-2) foi reportado em Dezembro de 2019, fato que foi oficialmente anunciado pela OMS no dia 9 de Janeiro. Incrivelmente, já no dia 12 de Janeiro foram obtidas as primeiras sequências genômicas do vírus, e assim começou a corrida para o desenvolvimento de vacinas.
Todos sabiam que este era um caminho longo, que normalmente demanda vários anos de desenvolvimento e estudos clínicos, antes de que uma nova vacina possa ser aprovada para uso na população. Por outro lado, existem inúmeros antivirais e outros medicamentos já aprovados para utilização em humanos, que poderiam ser mais rapidamente adaptados ("repurposed") caso demonstrassem atividade inibitória contra o SARS-CoV-2. Este caminho parecia ainda mais garantido com a rápida determinação estrutural de múltiplas proteínas do vírus, ainda durante os primeiros meses da pandemia (Protease, Spike, RdRp, etc). Se iniciou assim uma outra corrida, para a triagem virtual e testagem experimental de potenciais inibidores farmacológicos destas proteínas. Diversos estudos computacionais foram realizados, alguns testando milhares ou até milhões de compostos. Infelizmente, não se encontrou uma droga que conseguisse mudar o curso da pandemia. O melhor resultado foi obtido com o Remdesivir, que parece contribuir moderadamente para a redução do tempo de internação dos pacientes.
Felizmente os esforços na área do desenvolvimento de vacinas deram frutos, e muitos países começaram campanhas de vacinação já em Dezembro de 2020. Mas o que deu errado nos esforços de triagem virtual? O que ficou faltando? Alguns indícios apontam que na pressa por testar o maior número de ligantes possíveis, os pesquisadores abriram mão de considerar a flexibilidade das proteínas do vírus. Este tópico foi discutido por Kneller e colaboradores em um artigo publicado em Dezembro de 2020. Ao utilizarem uma nova técnica de cristalografia de raios-X na qual as estruturas são mantidas a uma temperatura mais próxima da temperatura ambiente, os autores observaram grande maleabilidade no sítio catalítico da enzima. Isso permitiu acomodar ligantes muito maiores do que se achava possível considerando o volume da cavidade revelada por métodos convencionais, incluindo a ligação de peptídeos (ou peptidomiméticos) com capacidade antiviral. Esta flexibilidade da proteína já havia sido sugerida em um estudo anterior, utilizando simulações de dinâmica molecular. Este tópico também foi brevemente abordado por mim em episódio recente do "Bioinformatics and Beyond Podcast", com Leo Elworth.
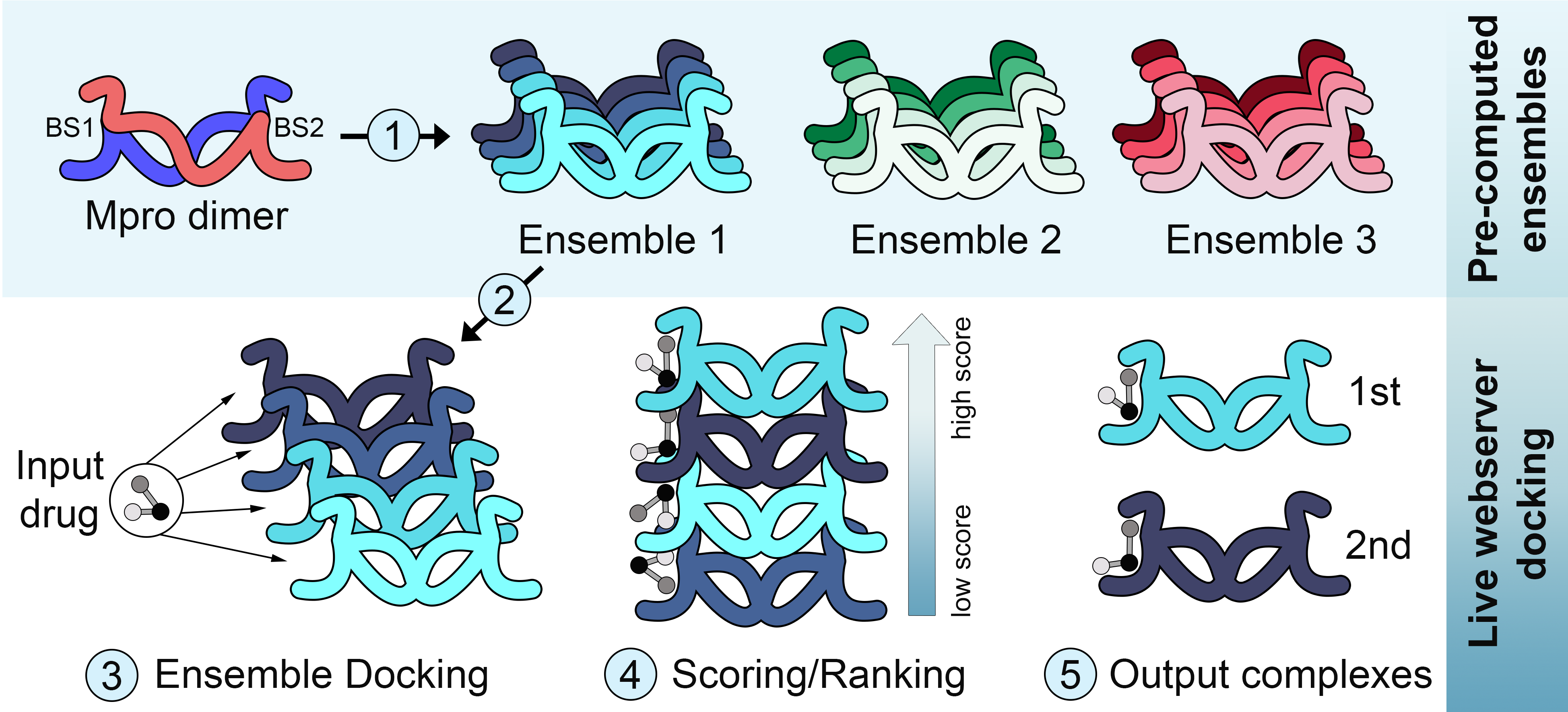
A maioria dos métodos de ancoramento molecular (docking) focam mais em explorar a flexibilidade do ligante, do que a do receptor. E sobretudo quando se deseja triar uma grande biblioteca de ligantes, a necessidade de se fazer predições rápidas torna praticamente inviável considerar a flexibilidade do receptor. No outro extremo, métodos como dinâmica molecular podem explorar a flexibilidade do receptor sozinho ou ligado a uma droga, mas tem um custo computacional que impede seu uso em larga escala. Uma alternativa intermediária é o chamado “ensemble docking”, no qual a flexibilidade do receptor é considerada de maneira implícita, ao se usar como input múltiplas conformações do receptor. Mas estes métodos apresentam um outro desafio: Como construir um ensemble que seja representativo dos estados conformacionais da proteína?
Ainda no primeiro semestre de 2020 eu fui abordado pelo Prof. Dr. Geancarlo Zanatta, da Universidade Federal do Ceará, sobre seu interesse em desenvolver métodos de ensemble docking para proteínas do SARS-CoV-2. O Prof. Zanatta começou a realizar múltiplas dinâmicas moleculares de proteínas do novo coronavirus, enquanto a nossa equipe começou a implementar um servidor para ensemble docking. Para o ancoramento, nós utilizamos um método paralelizado de meta-docking chamado DINC, previamente desenvolvido pela equipe da Prof. Dra. Lydia Kavraki da Rice University. Uma vez implementado o servidor e realizadas as simulações, um bom tempo foi dedicado ao desafio de selecionar um ensemble de conformações representativo para cada proteína do vírus. Para isso nós utilizamos uma combinação de métodos de redução da dimensionalidade (e.g., análise de componentes principais) e de clustering (e.g., k-means).
Estes métodos foram aplicados para construir ensembles para a protease principal (Mpro), a protease semelhante a papaina (PLpro) e a RNA polimerase dependente de RNA (RdRP). Os parâmetros e protocolos da simulação de dinâmica molecular tem impacto direto na amplitude das mudanças conformacionais observadas, e também existe variação entre replicatas de simulações. Para considerar o impacto destes fatores, o professor Zanatta utilizou dois protocolos distintos (ex., usando campos de força alternativos) e realizou múltiplas simulações com cada protocolo. Isso permitiu gerar mais de um ensemble de conformações para cada proteína, cada um explorando diferentes graus de variação em relação a referência cristalográfica. Além disso, para os casos em que havia um grande número de estruturas cristalográficas, um ensemble de estruturas determinadas experimentalmente também foi criado e disponibilizado (Figura 1).
Todos estes ensembles e o método de ensemble-docking já estão disponíveis online através do servidor DINC-COVID. O artigo descrevendo o desenvolvimento e implementação do servidor ainda está sob revisão, mas uma versão pré-publicação já está disponível no BioRxiv. Além disso, a equipe do professor Zanatta continua rodando simulações de outras proteínas de interesse, como a proteína S, as quais serão posteriormente adicionadas ao servidor. No vídeo abaixo, o Dr. Maurício Rigo, da equipe da Professora Kavraki, resume o projeto e descreve as principais funcionalidades do servidor.
Note que uma vez gerados os dados estruturais de uma proteina, seja por dinâmica molecular ou uma coleção de estruturas determinadas experimentalmente, a etapa de criação do ensemble para docking pode focar em diferentes partes da proteína. No caso da Mpro, por exemplo, nós geramos tanto ensembles para docking no sítio catalítico, quanto ensembles para docking em um sítio alostérico. Outra característica importante desta pesquisa é que a tecnologia desenvolvida neste trabalho poderá ser aplicada em outras áreas de investigação farmacológica, além do SARS-CoV-2, uma vez que as dificuldades encontradas na produção de ensembles conformacionais representativos de estados protéicos são comuns em muitos projetos. De fato, nossa equipe desenvolveu os protocolos de seleção das conformações utilizando pacotes públicos em python, e organizou as etapas em uma pipeline de fácil execução utilizando Jupyter Notebooks. A validação desta pipeline e os arquivos para rodar estas análises em outros sistemas estão sendo preparados para divulgação em uma segunda publicação.





- Apaixonados por Imunologia
- Comunicado
- Conteúdo Publicitário
- Curso
- Dept. Imunologia Clínica
- Dia da Imunologia
- Dia Internacional da Imunologia
- Divulgação científica
- Edital
- Especial
- Especial Dia da Imunologia
- Especial Doença de Chagas
- Evento
- Eventos
- Exposição COVID-19 da SBI
- História da Imunologia no Brasil
- Homenagem
- Immuno 2018
- Immuno2019
- Immuno2021
- Immuno2022
- Immuno2023
- Immuno2025
- Immuno2026
- IMMUNOLAC
- Immunometabolism2022
- Imune
- Imune - o podcast da SBI
- ImunoWebinar
- INCT Imuno
- Institucional
- IUIS
- Luto
- NeuroImmunology 2024
- Nota
- Nota Técnica
- Notícia
- o podcast da SBI
- Oportunidades
- Outros
- Parecer Científico
- Pesquisa
- Pint of Science 2019
- Pint of Science 2020
- Política Científica
- Sars-CoV-2
- SBI 50 ANOS
- SBI.ImunoTalks
- Sem categoria
- Simpósio
- SNCT 2020
- SNCT 2022
- Webinar
- WORKSHOP


