.png)

Autor: Dinler Amaral Antunes é professor assistente de Biologia Computacional no Departamento de Biologia e Bioquímica da Universidade de Houston, membro do Centro para Receptores Nucleares e Sinalização Celular (CNRCS), e da Sociedade Internacional de Biologia Computacional (ISCB).
O problema da predição de estruturas tridimensionais para proteínas monoméricas (ou seja, cadeia única) está resolvido!!! Este foi anúncio feito pela coordenação do CASP14 durante a cerimônia de apresentação dos resultados da competição, em Outubro de 2020. Se você trabalha (ou pretende trabalhar) nas áreas de Biologia Computacional e Bioinformática Estrutural, ou simplesmente acompanha notícias relacionadas a estas áreas, você provavelmente leu inúmeras manchetes sensacionalistas no final do ano passado, “A Google resolveu um desafio de 50 anos!”, “AlphaFold2 vai revolucionar a pesquisa em Biologia e Biomedicina”, “Biólogos estruturais ficaram obsoletos e precisam mudar de área de pesquisa”.

Todo este hype foi causado pela performance realmente impressionante do método chamado AlfaFold2 na última edição da competição bianual do CASP. O método foi desenvolvido por uma companhia subsidiária da Google, chamada DeepMind, a qual vem construindo um histórico de conquistas marcantes no uso de inteligência artificial para resolver os mais diversos problemas. Talvez você lembre de notícias sobre a performance de AlphaGo jogando Go, e de AlphaZero jogando Xadrez (sem ter “aprendido” xadrez).
Pois agora a história continua com AlphaFold (CASP13) e AlphaFold2 (CASP14). Seguindo tendências na área, o AlphaFold2 se utiliza de redes neurais e alinhamento múltiplo de sequências para predizer a estrutura tridimensional de uma proteína, a partir de sua sequência linear de aminoácidos. Esta é uma estratégia muito interessante para contornar um problema que limita a aplicação de métodos de inteligência artificial para muitas áreas de interesse em Biologia e Biomedicina, ou seja, a falta de dados anotados em volume suficiente para treinar um método de aprendizado de máquina (supervised machine learning approaches). No contexto de modelagem estrutural, por exemplo, o “dado anotado” seria uma proteína para a qual nós temos tanto a sequência de aminoácidos quanto a estrutura tridimensional determinada por métodos experimentais, como a cristalografia de raios-X. Se você pensou que este dado já existe, você está certo. O Protein Data Bank (PDB) tem mais de 100 mil proteínas com estrutura 3D determinada. Mas enquanto isso é um universo gigantesco do ponto de vista dos Biólogos estruturais, isso ainda é muito pouco do ponto de vista dos Biólogos computacionais e cientistas da computação tentando usar métodos de inteligência artificial. Os datasets usados para treinar redes neurais usadas no reconhecimento de imagens, por exemplo, tem milhares de entradas anotadas. Dados de sequências de proteína, por outro lado, são muito mais abundantes. Mais do que isso, ao se fazer o alinhamento múltiplo de sequências filogeneticamente relacionadas é possível se detectar correlações evolutivas entre resíduos em diferentes posições da proteína. Aminoácidos que evoluem/mutam de forma correlacionada, podem indicar restrições estruturais (note que a estrutura das proteínas está diretamente relacionada com sua função, o que acarreta maior conservação da estrutura/enovelamento do que da sequência linear de aminoácidos). AlphaFold2 utiliza uma nova arquitetura de inteligência artificial que permite a rede neural identificar quais pares de resíduos estão evoluindo de maneira correlacionada, quais destas possíveis interações são mais importantes, e até mesmo quais sequências no alinhamento múltiplo são mais relevantes. Isso tudo é feito em múltiplas rodadas de refinamento incremental, nas quais uma predição inicial é avaliada, e este “resultado” é utilizado como entrada para refinar todas as etapas do processo, do alinhamento múltiplo a estrutura 3D (veja artigo na revista Nature).
O impacto do AlphaFold2 para a área da Biologia estrutural é real e imediato. Ele alcançou uma média de 92.4 de GDT (Global Distance Test) considerando todos os desafios do CASP14, o que significa que na maioria dos casos ele conseguiu uma acurácia em nível atômico para a maior parte da proteína, mesmo em casos para os quais não havia nenhum molde disponível para guiar a predição (ex., ausência de proteína homóloga para a qual existe estrutura conhecida). O time da DeepMind também fez uma parceria com a EMBL para disponibilizar um banco de dados de proteínas modeladas (veja artigo na revista Nature). O objetivo ambicioso é modelar todas as proteínas conhecidas, e o banco já cobre cerca de 98% das proteínas humanas (~20 mil estruturas). Mais do que isso, a apresentação dos resultados impressionantes durante a CASP14 também influenciou os outros grupos competidores e acelerou a pesquisa na área. Para citar um exemplo, um outro time acaba de publicar outra ferramenta open source inspirada na arquitetura do AlphaFold2 (veja artigo na revista Science). Apesar de não superar o método original na modelagem de monômeros, o RoseTTAFold possui uma inovação que permite a modelagem de multimeros; potencialmente resolvendo uma das limitações do AlphaFol2, e expandindo a aplicação destes métodos para vários outros problemas em Biologia.
Em primeira vista, este salto de qualidade em relação a predição automatizada e altamente precisa parece representar uma competição desleal para biólogos estruturais, sobretudo considerando que a determinação de uma única estrutura por cristalografia de raio-x pode levar vários anos e render uma tese de Doutorado. Mais do que isso, mesmo a modelagem por homologia e a validação do modelo podem também exigir anos de trabalho e quem sabe render uma dissertação de Mestrado. Mas a situação no curto prazo pode ser justamente o oposto. A impossibilidade de se obter e analisar dados em larga escala tem impedido a biologia estrutural de alcançar um protagonismo equivalente ao das análises genômicas. Esta limitação permanece a despeito do fato de que em muitos casos o dado estrutural é essencial para se compreender a função da proteína em situações fisiológicas ou patológicas, e é também essencial para o desenvolvimento de fármacos. A facilitação do acesso a dados estruturais promovida por estas novas metodologias de modelagem pode alavancar esta área de pesquisa e criar novas oportunidades de trabalho. Por exemplo, estes métodos já estão sendo usados para se refinar dados experimentais, maximizando a informação que se pode extrair, e possivelmente acelerando o processo determinação experimental de outros alvos ainda mais complexos (veja artigos nas revistas Nature e Science). Além disso, existem muitos outros desafios computacionais para análise e validação destes modelos em diversos contextos, incluindo farmacogenômica e medicina personalizada.
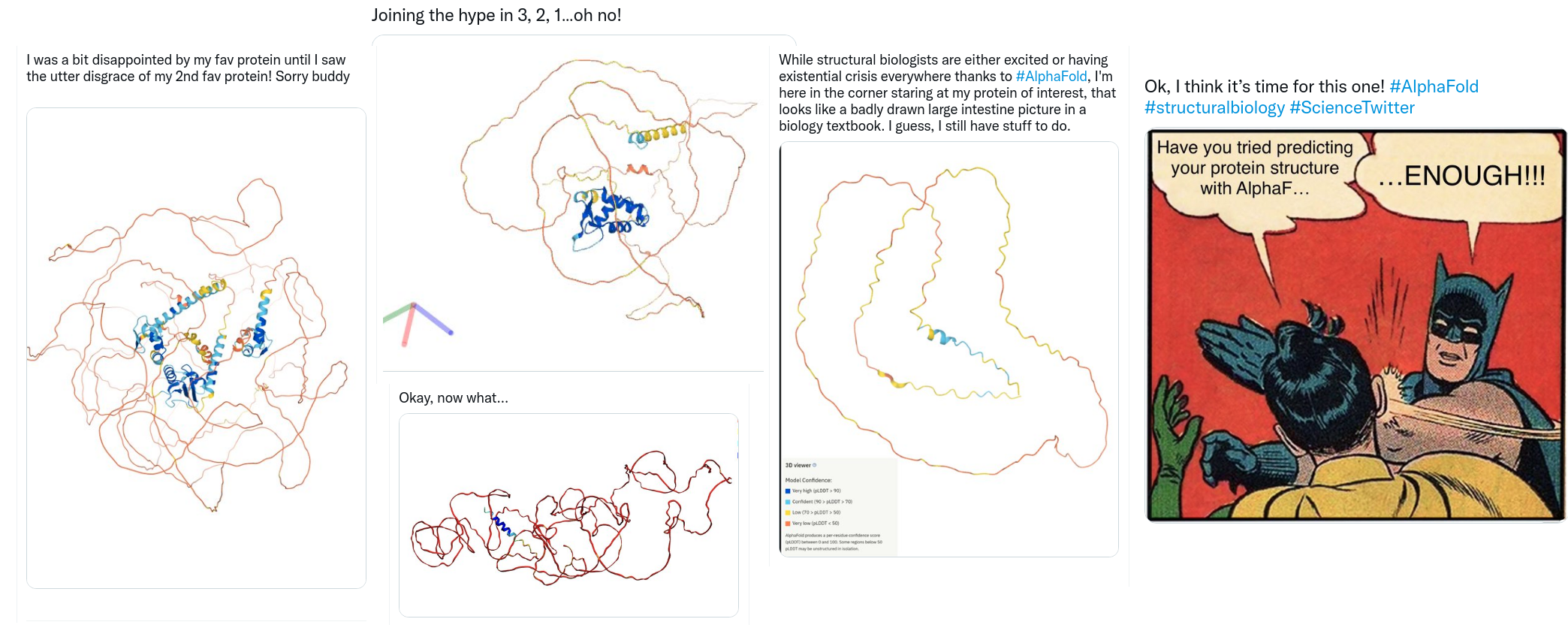
Finalmente, é importante salientar que ainda existem inúmeros problemas em Biologia Computacional e Estrutural que ainda não podem ser resolvidos com estes novos métodos. Por exemplo, grande parte do poder preditivo de métodos como AlphaFold2 e RoseTTAFold são derivados do alinhamento múltiplo de sequências e de inferências evolutivas. No entanto, existem proteínas de grande interesse biomédico para as quais não existem sequências relacionadas, ou as sequências relacionadas não contém informação evolutiva. Isso inclui por exemplo a modelagem estrutural de anticorpos, de receptores de células T, e de uma ampla gama de proteínas sintéticas ou derivadas de engenharia molecular. Finalmente, também e interessante observar que o banco de dados criado pela DeepMind em parceria com o EMBL contém inúmeras proteínas para as quais apenas uma pequena porção/domínio foi corretamente modelada (enquanto o restante é apresentado de modo completamente não enovelado). Isso gerou uma série de piadas e comentários nas redes sociais, nas quais pesquisadores se frustram (ou se divertem) com o fato de que seu alvo de interesse ainda não foi corretamente determinado. Portanto não se preocupe! Os métodos de inteligência artificial estão revolucionando esta área, mas a compreensão de aspectos biológicos (e o trabalho de Biólogos e Bioinformatas) ainda continua sendo indispensável para nos beneficiarmos ao máximo desta revolução.
Recursos úteis:
AlphaFold Database with EMBL-EBI:
https://alphafold.ebi.ac.uk/
AlphaFold on Github:
https://github.com/deepmind/alphafold
RoseTTAFold on Github:
https://github.com/RosettaCommons/RoseTTAFold
Google colab notebook para rodar modelagem com AlphaFold:
https://colab.research.google.com/drive/1LVPSOf4L502F21RWBmYJJYYLDlOU2NTL
Tutorial sobre como rodar AlphaFold utilizando Google Colab:
https://youtu.be/Rfw7thgGTwI




- Apaixonados por Imunologia
- Comunicado
- Conteúdo Publicitário
- Curso
- Dept. Imunologia Clínica
- Dia da Imunologia
- Dia Internacional da Imunologia
- Divulgação científica
- Edital
- Especial
- Especial Dia da Imunologia
- Especial Doença de Chagas
- Evento
- Eventos
- Exposição COVID-19 da SBI
- História da Imunologia no Brasil
- Homenagem
- Immuno 2018
- Immuno2019
- Immuno2021
- Immuno2022
- Immuno2023
- Immuno2025
- Immuno2026
- IMMUNOLAC
- Immunometabolism2022
- Imune
- Imune - o podcast da SBI
- ImunoWebinar
- INCT Imuno
- Institucional
- IUIS
- Luto
- NeuroImmunology 2024
- Nota
- Nota Técnica
- Notícia
- o podcast da SBI
- Oportunidades
- Outros
- Parecer Científico
- Pesquisa
- Pint of Science 2019
- Pint of Science 2020
- Política Científica
- Sars-CoV-2
- SBI 50 ANOS
- SBI.ImunoTalks
- Sem categoria
- Simpósio
- SNCT 2020
- SNCT 2022
- Webinar
- WORKSHOP


